Pancreatic Cancer Metabolism
Role of Autophagy in Pancreatic Cancer
Autophagy ("self cannibalism") is a process of cellular self digestion where a cell can digest portions of its cytoplasm, proteins, or even whole organelles to provide energy or cellular building blocks to allow the cell to survive times of stress. Importantly, we identified that pancreatic cancers have constitutively activated basal autophagy which they rely on for continued proliferation through its importance in the metabolism of these tumors (Yang et al., G&D 2011; Yang et al Cancer Disc 2014, 2018; Mukhopadhyay et al. PNAS 2021). This work has provided fundamental insights into the importance of autophagy in Ras-driven cancers and has motivated the opening of multiple clinical trials in pancreatic cancer using hyrdroxychloroquine as an autophagy inhibitor. Two recent randomized trials have shown that hydroxychloroquine increases the response rate in patients when combined with chemotherapy, demonstrating that it has activity in this disease. Current efforts are underway to understand the role autophagy in pancreatic cancer metabolism and to identify critical biomarkers of response to anti-autophagy therapies. We have also shown that autophagy allows pancreatic cancers to proliferate in the setting of DNA damage and therefore it may play a role in the intense therapeutic resistance of these tumors. To investigate these and other critical questions regarding the biology of autophagy, we are using a combination of in vitro and in vivo approaches using sophisticated mouse models of the disease.
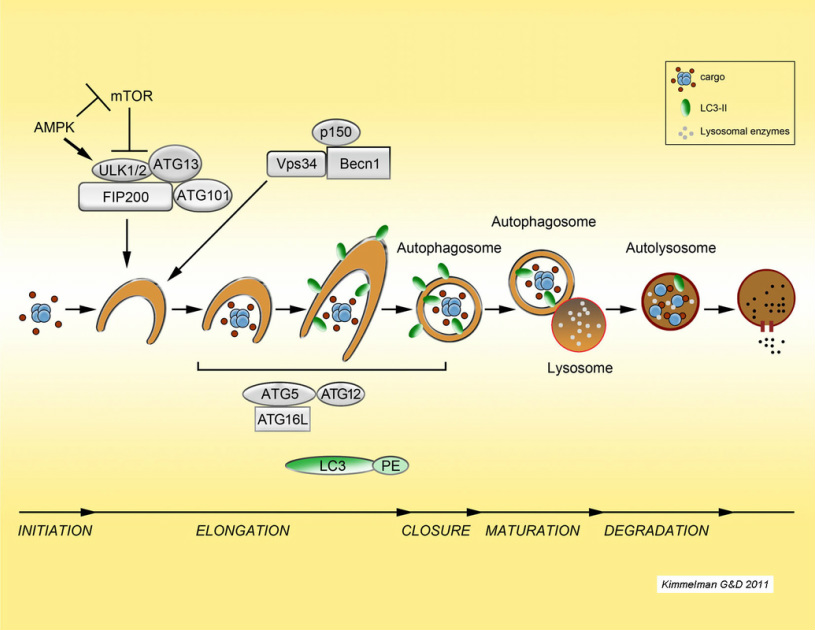
The complex process of autophagy
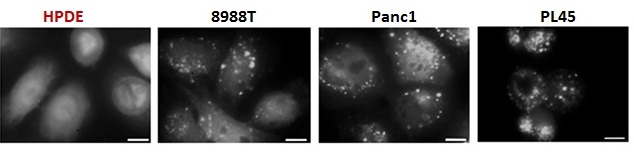
Pancreatic cancer cells have elevated autophagy, shown here by increased LC3 puncta compared to immortalized human pancreatic ductal cells (HPDE)
Autophagy promotes immune evasion of pancreatic cancer
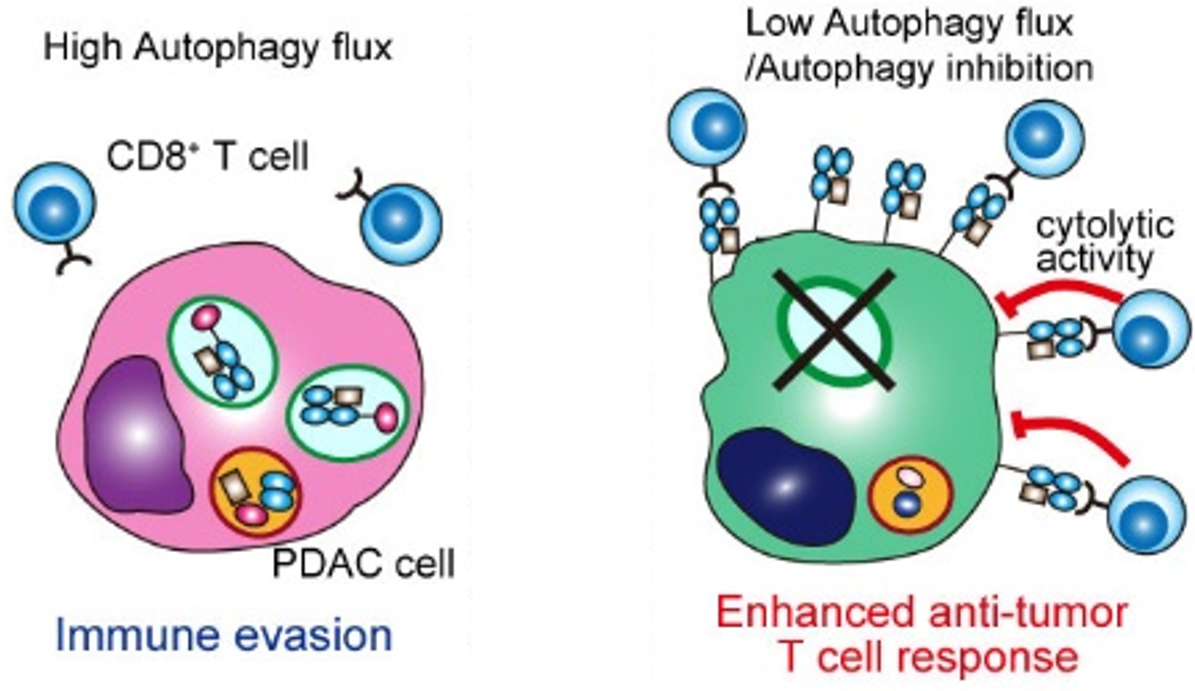
In collaboration with the Perera lab (UCSF), we identified a novel form of selective autophagy that degrades MHC-I (Yamamoto, Venida, et al. Nature, 2020). Therefore, the high basal levels of autophagy in these tumors cause them to have low levels of cell surface MHC-I, promoting immune evasion. These findings provide an explanation for the profound resistance of pancreatic cancer to immunotherapy. Importantly, we demonstrated that inhibition of autophagy increases infiltrating CD8+ T cells and synergizes with immune checkpoint blockade. We are currently working on further understanding the interplay of the complex immune response upon autophagy inhibition.
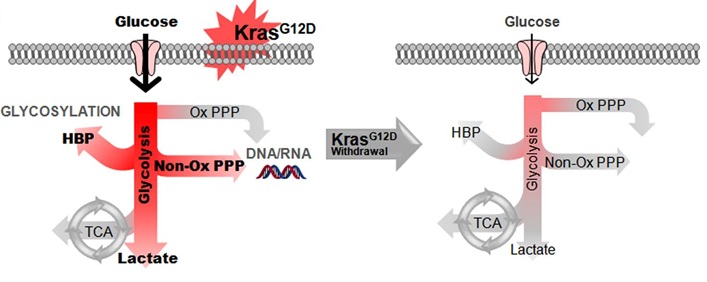
Kras Promotes the metabolic rewiring of Pancreatic Cancer
Using mouse models of pancreatic cancer where the Kras oncogene can be turned on and off, we were able to demonstrate that pancreatic tumors require the continued expression of oncogenic Kras for growth. Importantly, we identified that Kras has a critical role in re-wiring anabolic glucose metabolism (Ying et al., Cell 2012). In particular, it controls the rate limiting enzymes of the hexosamine biosynthesis pathway (HBP) and the non-oxidative arm of the pentose phosphate pathway (PPP). These are critical for protein glycosylation and nucleotide biosynthesis repectively. Furthermore, inhibition of the rate limiting enzymes of the HBP and the non-oxidative PPP lead to tumor regression.
Other work from our lab has demonstrated that pancreatic cancers utilize Glutamine via a novel pathway that is critical for redox balance (Son et al, Nature 2013).
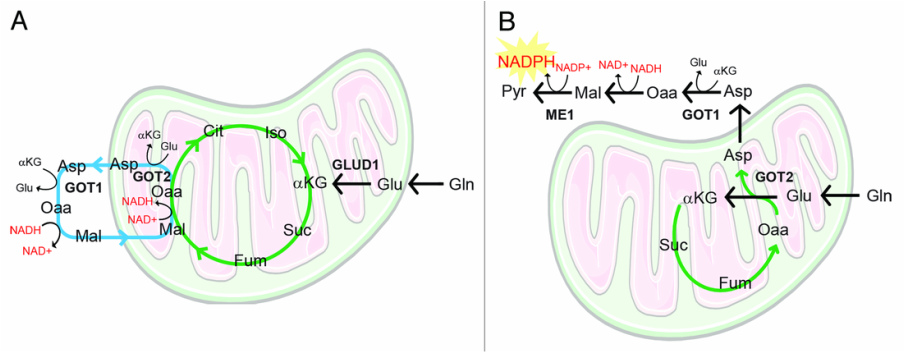
glutamine metabolism in pancreatic cancer via a novel pathway (panel B)
Current efforts are underway in the lab to understand how these tumors use various fuel sources utilizing a combination of metabolic labeling experiments and CRISPR/RNAi approaches in vitro and in vivo
Exploring the complex metabolic relationships between pancreatic cancer cells and the stromal microenvironment
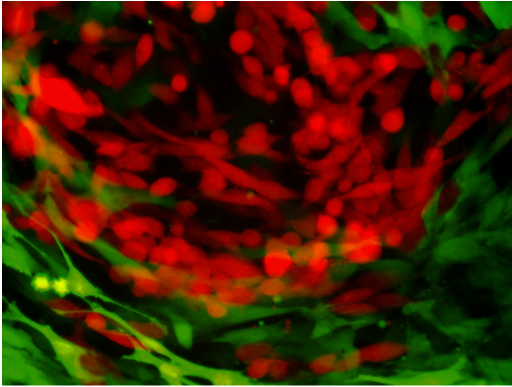
Pancreatic Stellate cells (GFP) can provide fuel sources to Pancreatic Cancer Cells (RFP)
We have previously demonstrated a novel metabolic cross-talk between pancreatic cancer cells and pancreatic stellate cells (Sousa et al., Nature 2016). Stellate cells can promote increased mitochondrial metabolism in pancreatic tumor cells. Through a series of experiments, we identified that this is due to the stellate cells secreting the amino acid alanine. Interestingly, tumor cells can signal to the stellate cells to increase autophagy. This promotes the degradation of protein, leading to the secretion of amino acids, in particular alanine. We demonstrate that the tumor cells take up alanine and use it to fuel the TCA cycle. This additional fuel source allows traditional fuel sources such as glucose to be used for other biosynthetic reactions such as to make nucleotides. Autophagy inhibition in the stellate cells themselves can disrupt this metabolic cross-talk and impair the growth promoting effects of stellate cells. More recently, we have explored the critical metabolic contribution of alanine to pancreatic cancer metabolism. We have identified the key transporters involved with alanine uptake and secretion and have shown that genetic deletion of the alanine importer (SLC38A2) in pancreatic cancer cells results in a metabolic crises and impaired tumor growth (Parker et al., Cancer Discov 2020). Further work into targeting nutrient cross-talk in the tumor microenvironment continues.
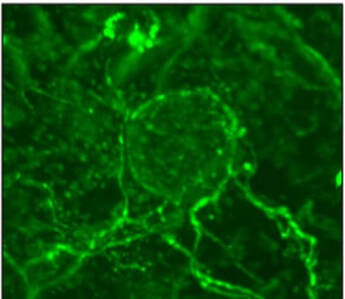
Nerves supply Pancreatic cancers with serine
In a recent study, we identified that neuronal innervation, a phenomenon commonly seen in pancreatic cancer, can funnel serine from the nutrient-rich peripheral circulation to the nutrient poor tumor microenvironment (Banh et al. Cell 2020). Interestingly, under serine-deprived conditions, pancreatic cancer cells secrete nerve growth factor through a program orchestrated by differential translation efficiencies of serine codons. Prevention of neuronal innervation using inhibitors to nerve growth factor receptor can impair tumor growth. Current work is focused on further exploring this fundamental biology as well as how this could be translated into effective therapeutic approaches.
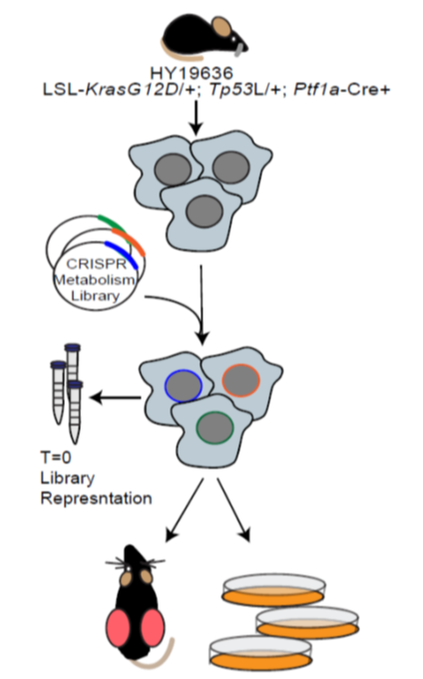
Schematic of in vitro and in vivo metabolism screens
In collaboration with the Aguirre lab (DFCI/Broad) we performed a series of in vitro and in vivo CRISPR screens using a custom library of metabolism genes (Biancur et al. 2021, Cell Metab). This work showed that in vitro conditions accurately model the majority of metabolic dependencies. Importantly, we identified Squalene synthase (FDFT1) as a potential therapeutic target in pancreatic cancer. Work continues evaluating metabolic dependencies, as well as using functional genomic approaches to study metabolism in pancreatic cancer.
